Platform Technology Funding
Project title: High-throughput genome-wide genetic screening using CRISPR/Cas9
Principle investigator: Dong-Yan Jin (School of Biomedical Sciences)
Co-investigator: Kin-Hang Kok (Microbiology), Man-Lung Yeung (Microbiology), Pengtao Liu (School of Biomedical Sciences)
Objective of the proposal:
In this project, we will develop the platform technology of high-throughput genome-wide genetic screening using CRISPR/Cas9-based RNA-guided genome editing. The use of CRISPR/Cas9 to knockout target genes in cultured cells or stem cells has been increasingly common in biomedical research. This technology has won the Tang Prize and the Alpert Prize, and it is widely predicted to get a Nobel Prize soon. In this general background, CRISPR/Cas9 screening is an emerging and “game-changing” technology platform that enables high-throughput functional screening at the genome scale using CRISPR/Cas9. Each of the known genes expressed in the target cell is specifically knocked out by use of CRISPR/Cas9-mediated genome editing. Based on the phenotypic readout (survival or death in most cases), all genes that affect a biological process can be identified. The common practice is to introduce a pooled single guide RNA (sgRNA) library into the cells and decode the sgRNAs using next-generation sequencing. It has been widely used in discovery research, interrogation of disease mechanism, drug target identification and rational design of precision therapy. This is an upstream platform but it is also useful in translational research. Currently it is not available in HKU and no one here has performed a CRISPR/Cas9 screen successfully.
There are three specific aims in this project:
1) Acquiring and optimizing the platform technology of CRISPR/Cas9 screening
2) Expanding the applications of CRISPR/Cas9 screening in biomedical research
3) Promoting and supporting the use of CRISPR/Cas9 screening in HKU and Hong Kong
Significance of the Proposed Platform Technology:
It was the dream of molecular genetists for many decades to knockout each gene completely and screen for the phenotype systematically. CRISPR/Cas9 screening is the emerging technology platform that makes this dream come true. The use of CRISPR/Cas9 in forward genetic screens has revolutionized functional genomics. Within a short time after its emergence in 2014, CRISPR/Cas9 screening has been successfully used in many different fields including molecular microbiology, cancer research, stem cell biology, cell and developmental biology, immunology, endocrinology, gastroenterology, neuroscience and drug discovery. It is a powerful tool with wide applications in biomedical science and represents a new era in functional screening. It is also rapidly evolving. It already has and will continue to have a great impact on molecular biology and medicine. CRISPR/Cas9 screening is not yet available at HKU, but it should be an integral part of many RF and TbRS programs in future. No matter we embrace it and invest early on it or not, CRISPR/Cas9 screening will be the next trend and it will become the new standard in biomedical research. Innovative use of this enabling technology platform in areas that we already have a competitive edge in the international arena will allow us to make new discoveries at an accelerated pace, to enhance our advantageous position, and to achieve the next level of research excellence. The technology platform can be used by anyone who has an interest in molecular biology and medicine. Therefore, a small investment on this enabling technology platform should have big returns.
Ongoing projects:
We have a long-term interest in studying Epstein-Barr virus (EBV). We have previously used CRISPR/Cas9 to edit Epstein-Barr virus genome for both mutant construction (Yuen et al.,J. Gen. Virol., 96:626-636, 2015; Yuen et al., Methods Mol. Biol., 1498:23-31, 2017) and virus eradication (Yuen et al., Virus Res., doi:10.1016/j.virusres.2017.04.019, 2017). To take one step further, we have applied CRISPR library screens in our EBV study. EBV is a herpesvirus that can switch from lytic replication to latent infection. EBV infects almost everyone but causes malignant cancer of lymphoid and epithelial origin in a small subset of people. Infection of nasopharyngeal epithelial cells with EBV causes nasopharyngeal carcinoma, which is particularly prevalent locally and in adjacent areas of China. In this study, we have carried out both positive and negative selection screens to shed light on host dependency and restriction factors that affect EBV lytic and latent infection in nasopharyngeal epithelial cells. Here are our feature projects:
1. Identification of the essential host dependency factors for EBV latent infection
A pooled sgRNA library on lentiviral vector was transduced into an immortalized nasopharyngeal epithelial cell line NP460 constitutively carrying GFP-EBV. The EBV in this line is in latency II state. Cell clones that have lost EBV can be sorted out by the loss of GFP with the help of flow cytometry (Fig. 1a). The knockout targets were then identified by next-generation sequencing. We have identified several candidates that are important for EBV latent infection (Fig. 1b). Our strategy is not only restricted to EBV study but can also apply to other latent viruses such as KSHV, HSV, HBV, etc. The only requirement is the virus needs to carry a fluorescent marker. The fluorescent marker can be inserted into the virus also by the CRISPR/Cas9 strategy. We have developed an efficient protocol to incorporate marker into the latent infected viral genome before (Yuen et al.,J. Gen. Virol., 96:626-636, 2015; Yuen et al., Methods Mol. Biol., 1498:23-31, 2017). By combining these two CRISPR-mediated methods, the essential host dependency factors can be identified on different latent infected viruses.
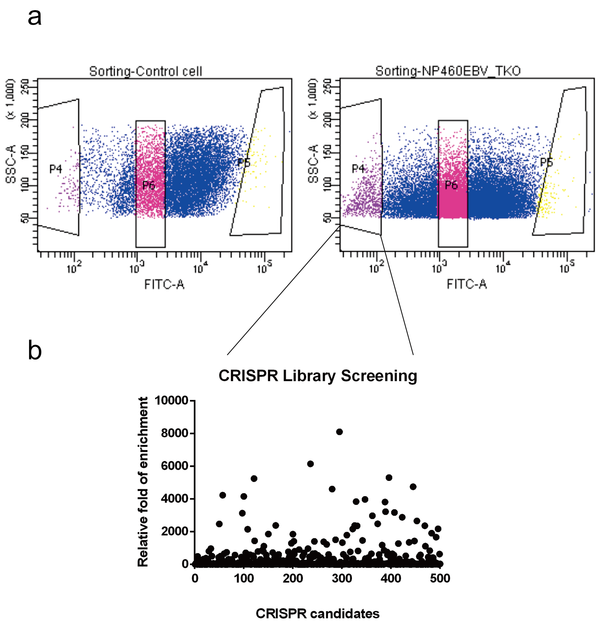
Fig.1 CRISPR library screening for essential EBV maintenance factors. a) A narrow GFP range of Nasopharyngeal epithelial NP460EBV cells (left panel P6) were sorted out by flow cytometry. These cells were challenged with the Toronto Knockout (TKO) CRISPR lentivirus library-version 3. Two days post-transduction, the TKO transduced cells were selected by puromycin and recovered for 5 more days. The EBV loss group (GFP -ve, right panel P4) was sorted out by flow cytometry. The EBV +ve group (GFP +ve, right panel P6) was sorted out as the control group. The gRNA containing cassettes in the sorted cells were amplified by 2 rounds of PCR and sent for MiSeq analysis. (b) Distribution plot for the top 500 CRISPR knockout candidates from the MiSeq results. Fold of enrichment was calculated by comparing the reads from the EBV loss group and the EBV positive control group.
2. Identification of the host dependency factors for EBV lytic reactivation
Other then the host dependency factors for EBV maintenance, cell host factors which can regulate the EBV latent to lytic switch can also be identified by the CRISPR library screening. To provide a selectable phenotype for EBV reactivation, we have established a series of EBV lytic sensitive fluorescent reporters for library screening (Fig. 2). We have applied the EBV lytic transcript promoters to drive the DsRed fluorescent marker expression. If the infected cells entered the lytic reactivation state after the knockout of the host regulators by CRISPR, the reporter plasmid will give the DsRed signal and we can make use of this signal to recover the target knockout cells by flow cytometry. In order to increase the sensitivity of the reporter, we have also constructed a new generation of reporters by using the positive feedback system. By the use of these sensitive reporters, we can identify the host regulators for the latent-lytic switch of EBV.
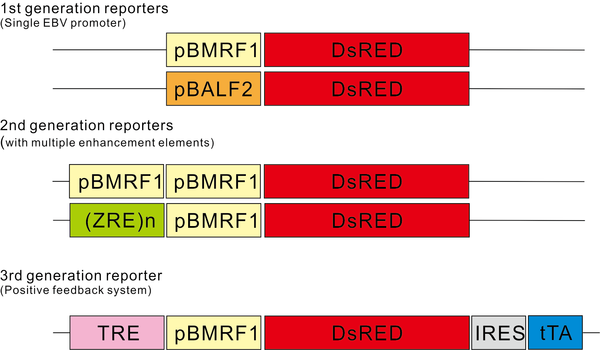
Fig.2 EBV lytic sensitive fluorescent reporters. 1st generation reporters: EBV lytic promoter pBMRF1 and pBALF2 were used to drive the DsRed expression. BMRF1 and BALF2 are among the most abundant EBV expressed proteins during lytic activation. 2nd generation reporters: Additional EBV lytic promoter or multiple copies of Zta response element (ZRE) were incorporated into the 1st generation reporter. 3rd generation reporter: A positive feedback loop was introduced into the 1st generation reporter using the tet-Off system. TRE (tetracycline Response Element) was added before the pBMRF1 and tTA was added along with IRES sequence downstream of DsRed. This design ensured that the initial expression of DsRed and tTA is driven only by pBMRF1. Once tTA is expressed in the system, it can activate TRE promoter to further express DsRed and tTA, thus establishing a positive feedback loop for higher expression of DsRed and increase the sensitivity of the reporter.
3. Identification of the functional partner of the viral proteins
Apart from the host dependency factors for the viral life cycle, CRISPR library screening can also be used to identify the functional partner of the viral proteins. Here we will use our Zta anti-interferon study to demonstrate how CRISPR library screening can be used to identify the unknown functional partner. Previously we have identified EBV protein Zta can potently suppress the interferon β (IFNβ) production, but the detailed mechanism as well as the host interaction partners are still unclear. To get some hints for further study, we have established a CRISPR library screening system to identify the host partners for Zta. We have constructed an IFNβ promoter-driven GFP fluorescent reporter and the Zta-IRES-DsRed expression construct for the library screening. In this system, HEK293 cells were stably transfected with a pIFNβ-GFP reporter plasmid. The GFP expression is strictly controlled by the IFNβ promoter and can only be expressed in the presence of activators such as RIG-I N shown in Fig. 3. In the presence of Zta, GFP expression was totally abolished due to its potent inhibitory effect on IFNβ production (Fig. 3). Based on this phenotype, we can combine this GFP reporter system with CRISPR knockout libraries to screen out cellular proteins essential for Zta-mediated suppression of IFNβ production. As the DsRed marker will be co-expressed with Zta under the control of the IRES polycistronic element, the GFP (indicating pIFNβ activity) and DsRed (indicating the presence of Zta expression vector) double-positive cells should not appear, unless the essential partner required for Zta-mediated suppression of IFNβ production is knocked out by CRISPR. The GFP-DsRed double- positive cells can then be isolated by flow cytometry and the knockout gene can be identified by next-generation sequencing. This approach is not only restricted in the virology study but also applies to any interaction which involved a third interaction partner.
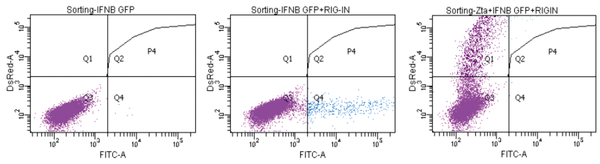
Fig. 3 Due-fluorescent reporter system for functional partner identification. Inhibition of the activation on IFNβ-GFP activity by RIG-I N. HEK293-IFNβ-GFP stably transfected cells were co-transfected with RIG-I N and Zta-IRES-DsRed as indicated. The cells were harvested 48 hours post-transfection and the percentage of GFP and DsRed positive cells were analyzed by flow cytometry.
Available CRISPR screening libraries:
Toronto KnockOut (TKO) CRISPR Library - Version 3
- 70,948 guides (4 guides/gene) targeting 18,053 protein-coding genes
- Lentiviral library (Puromycin resistance)
- Constructed by Moffat Lab and obtained from addgene (https://www.addgene.org/pooled-library/moffat-crispr-knockout-tkov3/)
Human CRISPR Activation Library (SAM - 2 plasmid system)
- 70,290 guides targeting 23,430 protein-coding genes
- lenti-SAMv2 (contains dCas9-VP64 and sgRNA, Blasticidin resistance)
- lenti-MPHv2 (contains MS2-P65-HSF1 activator helper, Hygromycin resistance)
- Constructed by Zhang Lab and obtained from addgene (https://www.addgene.org/pooled-library/zhang-human-sam-v2/)